
The pre-IND meeting is a unique opportunity to meet with the FDA to discuss questions about drug development program and to help ensure a complete IND application is submitted. The pre-IND meeting is free of cost and can help avoid clinical holds and costly missteps.
https://www.nuventra.com/resources

FDA와의 첫 만남
우리 회사 leading project인 NN2101의 임상1상 IND를 준비하면서, FDA에 regualtory document 제출을 처음으로 준비하게 되었다. Clinical hold는 어떤 상황에 먹을 수 있고, 어떻게 clear할 수 있는지, stability data가 어디까지 필요한지, IMP 레이블에 shelf-life를 표시해야 하는지, 그렇다면 재포장 과정이 필요할지 등 궁금한 점이 많았다. 가이드라인, consultant등 FDA 경험자들의 조언을 통해 어느정도 감은 잡을 수 있었지만, 그래도 해결되지 않는 부분을 FDA로부터 확인 받고 싶었다.
Pre-IND는 잘 활용한다면 스폰서의 전략을 IND 가기전에 FDA에 전달할 수 있는 특별한 기회이다. FDA가 pre-liminary answer를 통해서 혹은 미팅 중에 준 코멘트를 충실히 IND package에 반영하면 개발 전략이나 데이터에 대한 FDA와 sponsor간의 gap을 좁혀 clinical hold 리스크를 최소화 할 수 있을 것이다.
D-60
Meeting Request Submission
Type B 미팅의 경우 질문 리스트를 제출 하는 것에서 부터 시작된다. CMC, preclinical, clinical 파트별 질문을 많게는 4개 정도씩 총 10 개 내외로 작성 간결하게 작성해서 제출했다. 질문의 형식은 우리의 전략이나 데이터에 대해 설명하고 IND package 구성하기에 적합한 데이터라고 생각하는가로 물어보고 'yes' or 'no'로 답변 받을 수 있게 구성했다. 질문의 내용은 정해진 것이 없다. 임상1상을 위한 IND를 준비 중이라면, patient safety와 직결되는 DP stability data, preclinical data, starting dose, dose escalation strategy 등에 초점을 맞춰서 개발상황을 잘 반영한 내용으로 구성할 수 있다.
D-40
Meeting Request Granted from FDA
질문 리스트 제출 후, FDA 담당 regulatory project manager (RPM)이 배정되는데, 미팅 시간 조율을 위해 선호하는 시간을 서로 주고 받는다. 미팅 시간에 대한 합의가 되면 FDA로부터 공문을 받게 된다.

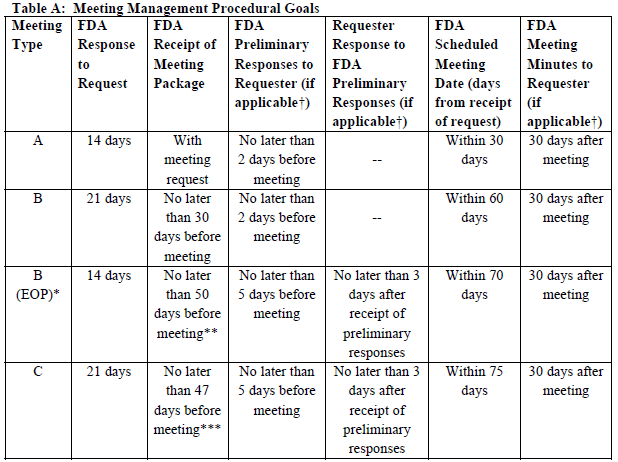
Type B의 경우 21일 안에 미팅 요청에 대한 회신을 FDA로부터 받게 된다. "Meeting Request Granted"라는 제목의 공문 형식으로 일정, 형식(face to face, teleconference, vedioconference 등), FDA측 미팅 참가자, briefing package 제출 기한 등에 대한 정보가 포함되어 있다.

D-30
Briefing Package Submission
너무 내용이 많지도, 그렇다고 적지도 않아야 한다. Meeting request를 통해 질의했던 질문들과 함께 뒷받침 하는 주요 데이터들을 테이블, 그래프 등을 활용하여 작성한 다음 미팅 30일 전까지 제출하면 된다. 제 때 제출하지 않으면, 미팅이 취소되거나 일정을 다시 잡아야 하기 때문에 기한을 지키는 것이 중요하다!
FDA CDER 관련 submission은 NextGen portal에서 이루어지며,briefing package를 제출 하고 나면 아래와 같은 화면이 나타난다. 꼭 필요한 절차는 아니지만, 담당 Regulatory Project Manager에게 제출 했음을 메일로 notify해주면 submission 잘 받았다고 컨펌을 받을 수 있다.

D-2
Preliminary Answer from FDA
Type B meeting의 경우 빠르면 미팅 이틀 전에 FDA preliminary comments를 받을 수 있다. General lead time이기 때문에 FDA 상황에 따라 반나절 전에 답변이 오는 경우도 있다고 한다.
Preliminary comments가 충분하다고 판단이 되면 미팅 취소 요청을 해도 되고, 해결되지 않은 부분이 있다면 미팅을 활용하여 FDA로부터 추가 설명을 들을 수 있다. Point delivery를 효과적으로 하기 위해, 확인이 필요한 부분 한 두 개를 topic으로 선정해서 집중적으로 논의 하는 것이 좋다. 필요에 따라 slide를 준비해서 미팅에 참여해도 되고, 미팅 후에 RMP에게 meeting minute과 함께 meeting material들을 제공해주면 된다. 단, meeting request나 briefing package 등 이전에 제출되었던 문서에서 벗어나지 않는 내용이어야 한다.
미팅 때는 이전에 제출 했던 질문들과 연관없는 내용이나, 새로운 질문은 언급하지 않는 편이 좋다. 그런 부분들은 미팅 요청을 다시 해서 따로 논의 하기 바란다.

D-0
Pre-IND meeting
총 60분간 진행되며, 최대 5분 동안 sponsor측 참가자들이 짧게 자기 소개를 하는 것으로 미팅이 시작된다. 우리는 slide deck을 준비해갔기 때문에, 준비한 자료를 띄워 놓고 준비해 간 질문을 했고 바로 FDA 답변을 받을 수 있었다. 또 FDA 측에서 종 선정 등 그들이 critical하다고 생각하는 부분에 대해서 comment를 줬고, 어떤 자료들을 준비해가면 좋을지 질문하고 답변받는 식으로 자연스럽게 대화가 오고 갔다. 최대 50분 간 discussion을 할 수 있는데, 이는 생각보다 짧다. 두 세개 토픽을 선정해 간다면 한 토픽당 길어야 15분 내외정도로 시간을 쓸 수 있기 때문에, 발언자를 정해놓는 등 사전 세팅을 깔끔하게 마무리 한 상태에서 미팅에 참여해야 한다. 마지막 5분 동안은 RMP이 meeting wrap up하는 시간을 갖고 끝난다.
Meeting minute은 FDA에서 작성하여 미팅 후 한달 이내로 공문형식으로 sponsor에게 전달하고, FDA서버에도 기록을 남겨두기 때문에 이후 IND 때 reviewer들이 참고하게 된다. Sponsor도 미팅 후에 meeting mintue을 제출 할 수 있는데, 어디까지나 참고용이므로 공식 meeting minute에 반영되지 않을 수도 있다.
FDA와 커뮤니케이션을 효율적으로 하기위해 참고하면 좋은 가이드라인
FDA와 미팅 시간은 매우 짧다. 미팅 목적이나 중요한 타임라인 등을 미리 파악할 수 있다면 미팅 준비를 철저하게 할 수 있을 것이다. FDA에 IND 등 제출할 계획이 있다면 우선 FDA와의 공식적인 커뮤니케이션에 대한 가이드라인을 확인해보기 바란다!
How-to-prepare-for-a-successful-meeting-with-CDER.pdf
0.18MB
Best Practices for Communication Between IND Sponsors and FDA During D
Procedural
www.fda.gov
Formal Meetings Between the FDA and Sponsors or Applicants of PDUFA Pr
Procedural
www.fda.gov
https://www.fda.gov/vaccines-blood-biologics/biologics-procedures-sopps/section-8100-communication
파트별 Pre-IND 미팅을 준비하는 방법
앞의 내용들은 Pre-IND meeting 준비에 대한 총론에 해당한다면, 지금부터는 미팅 전후에 알게된 사실들을 파트별로 정리해봤다. 처음 FDA와의 공식 미팅을 준비하는 분들께 도움이 되길 바란다.
Development Background/MoA
데이터에 기반한 이론적 설명
사실 IND에서 FDA main focus는 CMC와 safety 자료이다. 그래서 규제적 관점에서 MoA와 primary PD를 포함하는 initial efficacy study에 관한 정해진 규칙이란 없다. 질환에 대한 정보, 시험 물질을 약으로 개발하게된 배경, highlight하고 싶은 작용기전, 시험물질과 연관성을 보기 위해 수행했던 시험의 결과들을 regulator들의 공감을 살 수 있는 하나의 스토리라인으로 제시하면 된다. Supporting document로는 key study의 경우 raw data를 포함한 report를 제출하면 되며, protocol은 필수사항은 아니다. 이외 study는 논문을 reference로 제출하면 된다.
Preclinical
제품 특성을 고려한 종 선정과 human dose 설정
임상 1상을 가기위한 IND라면 toxicology, pharmacology study 등 safety 자료가 IND package의 메인이 될 것이다. 특히 이들 시험에 사용된 동물이 개발하고자 하는 제품의 안정성을 입증할 수 있는 종인지 종 선정에 대한 근거와 그를 뒷받침하는 충분한 데이터를 제시할 수 있어야 한다.
Human dose는 toxicology, pharmacology study 결과에 근거해서 산출하게 되는데 species relevancy, safety factor가 dose conversion strategy의 key라고 할 수있다.
Biotechnology 제품을 개발하고 있다면, 비임상 시험에 적합한 종 선정 시 아래 가이드라인을 참고해볼 수 있다.
S6 (R1) Preclinical Safety Evaluation of Biotechnology-Derived Pharmacueticals
CMC
IND에서 요구하는 최소한의 요구사항 준수
사실 임상1상을 위한 IND에서는 CMC 자료는 기본적인 요건만 충족한다면 pre-IND에서 critical한 comment는 받지 않고 지나갈 수 있다. 제일 궁금했던 부분은 안정성 데이터 요건이었는데, 일반적으로 IND에서는 clinical batch에 대한 release test 결과만 요구되며 나머지 안정성 데이터는 필수사항이 아니다. 주의해야 할 부분은, 만약 개발 과정에서 DP batch에 major change가 있는 경우(예-제조소 변경) batch 간 consistency를 입증해야 하며 clinical batch 1개월 안정성 데이터의 제출이 필요할 수 있다.
IND Applications for Clinical Investigations: CMC
FDA와 관련한 유용한 사이트
Search for FDA Guidance Document
CDER Manual of Policies & Procedures | MAPP
FDA-Approved Drugs
CFR - Code of Federal Regulations Title 21
노벨티노빌리티 RA팀 정규원
'Our Story' 카테고리의 다른 글
| [토르 망치] 노벨티노빌리티 사내 모임 지원제도 (0) | 2023.03.16 |
|---|---|
| 노벨티노빌리티 뉴스레터 창간 (0) | 2023.03.16 |
| 2022 노벨티노빌리티 야유회 (0) | 2023.03.16 |
| 결혼기념일 챙겨주는 회사가 있다!? (0) | 2023.03.16 |
| ARVO 2022에 다녀왔습니다. (0) | 2023.03.16 |
